The Promise and Puzzle of Scaling Laws
In natural language processing (NLP), scaling laws have given researchers a blueprint for how compute, parameters, and data can predictably affect model loss (Kaplan et al., 2020; Hoffmann et al., 2022). This predictability has transformed training from empirical guesswork to dial-tuning. In other words, once the scaling law has been established, the recipe is simple – pick a loss target, then back out the combination of compute, data and parameters that are required to meet that target using a few simple equations. Despite the promise that scaling laws have made in designing predictable model losses, when they have been applied to downstream functional tasks, they have led to puzzlingly unpredictable outcomes (Lourie et al., 2025).
Do NLP-styled scaling laws provide the same roadmap for biology?
Protein variant effect prediction now boasts dozens of models spanning architectures, sizes, and training corpora, however the progress of protein language models (pLMs) has slowed. This can be seen in ProteinGym (Notin et al., 2023) where the top score for protein variant effect prediction is a Spearman correlation of 0.518 (AIDO Protein-RAG, 16B), while the strongest pLM entry (ESM3) sits well below that in zero-shot substitution prediction (14th place with Spearman = 0.466), and the spread among pLMs is a modest 0.052. In other words: plenty of modeling diversity, surprisingly tight performance bands. That stall is exactly what prompted our question: Do the assumptions that make scaling work in NLP apply here? And can we use scaling laws that were developed for generalized pLMs to model empirical relationships in the downstream task of protein function prediction?
The Unruly Nature of Biological Data
Most scaling law studies in protein fitness have focused on the effects of optimizing compute and parameters (Cheng et al., 2024; Fournier et al., 2024; Lin et al., 2023), with far fewer exploring the stability of data distributions (Notin et al., 2022). This may be driven by the fact that biological data is messy and holds several unique challenges:
- Redundancy. Heavily overrepresented families and taxa dilute effective novelty. (Ding & Steinhardt, 2024; Poux et al., 2016).
- Noise. Sequence quality and preprocessing vary widely across sources (Chorlton et al., 2024).
- Sparse labels. Most sequences lack experimental validation, especially across multiple properties (Rauer et al., 2021).
- Functional multiplicity. Proteins can have multiple or context-dependent functions (Jeffery et al., 2023).
- Limited coverage. We’ve sequenced only a fraction of Earth’s proteins (Louca et al., 2019).
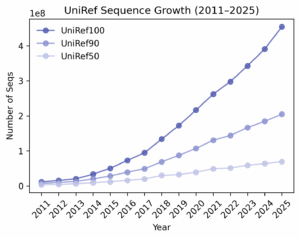
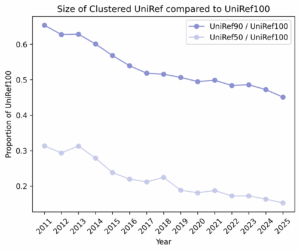
This leaves us with our core question: Does more unlabeled sequence data reliably improve pLM performance?
Unraveling the Problem: The AMPLIFY Experiment
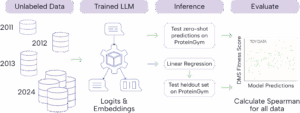
To explore whether more data has actually led to better pLMs, we turned to a new resource uniquely suited for isolating the effects of training data scale: the AMPLIFY suite of pLMs (Fournier et al., 2024). Each AMPLIFY model was trained on a cumulative yearly snapshot of UniRef100 (2011 -> 2024) using the same architecture and training setup. By holding everything else constant, the changes in performance can be directly evaluated in terms of data quantity and composition.
This approach allowed us to ask the question: Has the last 13 years of UniRef growth actually produced richer, more capable models? We approached answering this question in two ways:
- Predict the fitness of protein variants in the ProteinGym benchmark to see if models trained on more data do a better job at predicting variant effects.
- Use the learned embeddings from the model for simple supervised learning experiments to determine if embeddings from later timepoints contained information that improved predictions.
What We Found
Zero-shot AMPLIFY (2011–2024): TLDR; increase in performance is not monotonic.
Zero-shot Spearman correlations showed no steady climb, instead fluctuating year-to-year with dips even as billions of new sequences were added. Breaking results down by MSA depth (i.e. how many related natural sequence exist in UniProt) helped clarify this pattern: generally, proteins with higher MSA depth often improved with additional data, whereas proteins with low MSA depth sometimes performed worse with newer data.
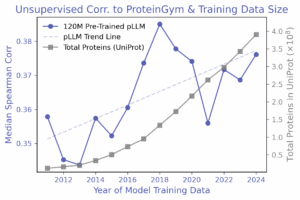
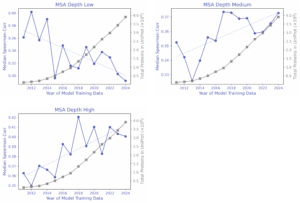
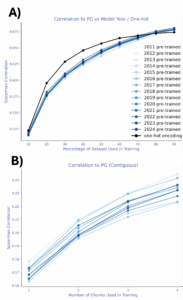
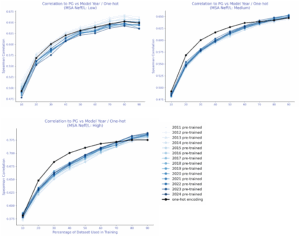
Supervised probes: TLDR; labels help with prediction, but leakage can lie.
A simple linear regression on a random 10% split with AMPLIFY embeddings, consistently outperformed zero-shot, and performance improved monotonically as more labeled data was added. Surprisingly, one-hot encodings beat embeddings from every AMPLIFY model in this setup-until data leakage was addressed: random train/test splits let the same mutational positions appear in both sets, inflating performance. With leakage-controlled splits—contiguous or modulo (from ProteinNPT; Notin et al., 2023)-scores dropped sharply, especially for one-hot encodings. No year-over-year trend was observed for AMPLIFY embeddings in either the leakage-prone or leakage-controlled settings.
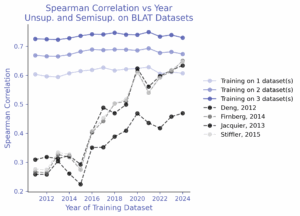
β-lactamase: TLDR; targeted datasets present a clearer pattern.
Finally, we focused on β-lactamase, the ProteinGym target with the most assays. Here, a clearer pattern emerged: zero-shot performance improved steadily with newer training data. AMPLIFY models trained on later years consistently outperformed earlier years. By contrast, semi-supervised learning told a different story: performance did not trend upward year-over-year. It remained flat. Training on one experimental dataset from 2011 matched the zero-shot accuracy of a 2020-trained model, and adding more β-lactamase datasets pushed performance beyond all unsupervised baselines.
Why didn’t scaling laws appear?
The assumptions behind smooth scaling didn’t hold. We likely haven’t hit data saturation for protein LMs: models remain sensitive to which sequences are added each year. In later UniRef100 snapshots, growth was dominated by redundant sequences, reflected in the declining UniRef90/UniRef100 diversity ratios – in general, the corpus got bigger without necessarily becoming more informative. That helps explain why low-MSA targets sometimes fared worse with newer models, while high-MSA targets typically fared better. Composition shifts-e.g., surges of viral proteins during COVID-19-may have tilted the training distribution in ways that don’t generalize across diverse families. Bottom line: our research suggests that composition and effective diversity matter more for year over year model performance than sheer size; without them, the tidy scaling curves don’t materialize.
Caveats Here for Understanding Data Scaling
Our study makes clear that if composition matters more than sheer size, then identifying which data drives performance is critical. In studying this, we uncovered two key issues:
- Using UniRef100 without redundancy reduction or sampling correction highlights just how much bias exists in the most widely used pretraining dataset—certain families and taxa are overrepresented to the point of skewing results.
- Using models trained with only a single random seed revealed how fragile year-to-year comparisons can be, with some fluctuations likely reflecting training variance rather than real biological signal.
Together, these findings show that current practices risk blending signal with noise, and point directly to what’s needed next: deduplicated or balanced sampling strategies, and multi-seed training runs that can distinguish variance from true scaling effects.
How to directly address these caveats
Our results show that simply scaling raw data isn’t enough; to understand how data truly shapes protein LMs, we need experiments that cleanly separate composition, size, and randomness while holding architecture and compute constant. This means:
- Build a temporal x redundancy grid.
Train identical models across years (2011→2024) x cluster thresholds (UniRef30/50/70/90). For each cell, log effective diversity (e.g., Neff/L), duplication rate, and taxonomic mix. This isolates the roles of sequence diversity and recency from raw token count. - Run multi-seed, fixed-budget replications.
Use 3–5 seeds per configuration (same steps/optimizer) to separate training variance from true data effects. Report the mean ± IQR so “improvements” aren’t just lucky runs. - Balance the mixture, not just the volume.
Apply balanced sampling across clusters/taxa (as in ESM2-style caps or reweighting) to prevent dominant groups from drowning out rare sequences. Compare this to an unbalanced baseline to quantify composition sensitivity. - Deduplicate systematically.
Train with no dedup, near-duplicate removal (e.g., identity caps), and aggressive dedup; report how performance tracks effective (not raw) tokens. This tests whether gains are from novel signals versus repeated sequences. - Attribute predictions back to data.
Use influence functions / TracIn / data-Shapley to trace downstream predictions to specific training cohorts. Remove or upweight those cohorts to see when and why scaling laws hold or fail. - Leakage-safe evaluation.
Use contiguous/modulo splits and homolog-aware partitions; include cross-protein generalization. Keep metrics identical across cells so curves are comparable. - Publish a transparent recipe.
Release dataset hashes, sampling rules, cluster thresholds, and seeds. A standard report (data stats → training logs → eval splits) turns ad-hoc observations into actionable principles.
The outcome to aim for: marginal-utility curves that show how much new performance each extra unit of effective diversity (not just tokens) buys you, and a compute-aware recipe for choosing which sequences to add next.
Broader Challenges for the Field
Our findings point beyond the specifics of UniRef and AMPLIFY to highlight broader challenges that the entire field will need to address if we want reliable scaling laws in biology:
- Redundancy vs. diversity. Large biological corpora are skewed toward a few organisms and families; curating datasets that grow and scale with effective diversity remains an open problem (e.g., balanced sampling, dedup at collection time, targeted long-tail acquisition).
- Label scarcity and narrow benchmarks. Experimental labels are limited and uneven and benchmarks like ProteinGym are invaluable but cover a thin slice of protein function space, making it hard to validate broad gains or capture context-dependent effects.
- Transfer and robustness. Models tuned on one family or task often fail to generalize across proteins, conditions, or assay types; we need evaluations that stress cross-protein transfer, distribution shift, and out of distribution behavior – not just in-family splits.
- Evaluation hygiene at scale. Preventing leakage (across mutations, positions, and homologs), handling batch/plate effects, and reporting uncertainty are prerequisites for trustworthy scaling curves, not optional extras.
- Data governance and provenance. Reproducible scaling requires stable data recipes (hashes, clustering thresholds, sampling rules) and transparent provenance so improvements can be traced, audited, and replicated.
Solving these will require coordinated work across data generation, curation, and evaluation design—not just “more data” or “bigger models.”
Where This Leaves Us…
Our results show that more data alone isn’t enough to unlock better protein language models: we believe the real driver will be composition and diversity. Year-over-year UniRef growth added billions of sequences, but without balancing redundancy or variance, performance stayed flat and sometimes even declined. What we need now are scaling studies that treat diversity as the currency of progress, separating true signal from noise and linking model predictions back to the data that shaped them. Beyond our work, the field must take on broader challenges: richer benchmarks, informative evaluation, and reproducible data recipes. Scaling laws gave NLP a blueprint; now it’s time to create that map for biology.